



CRISPR(クリスパー)ゲノム編集ソリューションズ
ベクタービルダー社の包括的なCRISPR製品とサービスはin vitroまたはin vivoゲノム編集を必要とする実験へ理想的なツールとなります。当社は、トランスフェクションやトランスダクションに使用する市販の試薬から、複数の納品形態(プラスミド、ウイルス、RNAなど)で提供される高ターゲティング効率をもつカスタムCRISPRベクターまで様々なCRISPR製品群を提供しています。トランスフェクションが難しい細胞に対してCRISPRを実行するために、主要なウイルスタイプ(レンチウイルス、AAV、アデノウイルスなど)へのパッケージングサービスも提供しています。さらに、当社はノックアウト、遺伝子発現活性化、遺伝子発現抑制、その他のCRISPRスクリーニング用途の高品質CRISPRライブラリー構築を専門としています。当社が独自に設計し、実用性が確認された全ゲノムデュアルgRNAノックアウトライブラリーは、ヒトとマウス遺伝子の機能スクリーニングへの強力なツールとなります。
特長
- 直感的に扱えるオンラインベクターデザインプラットホームと全ゲノムgRNAデータベースによって簡単で素早いCRISPRのデザインが可能です。
- 豊富なベクターバックボーンとコンポーネントが利用できます。
- 様々な納品形態(CRISPR/Cas9プラスミド, CRISPR/Cas9ウイルス, Cas9 mRNA + gRNA, Cas9-gRNA RNP複合体)を選択できます。
- 多様なCRISPRライブラリー構築オプションを選択できます。
- 信頼性の高い品質、短い作業日数、競争力のある価格。
- 実験計画の設計、データ解析、トラブルシューティングへの強力な技術サポート体制。
製品内容
- カスタムCRISPRベクター
- ポピュラーCRISPRベクター
- CRISPRウイルス
- Cas9 mRNAとgRNA用のRNA調製
- ゲノム編集用ドナーDNA
- CRISPRライブラリー
- CRISPRソリューションズ
技術情報
- CRISPRゲノム編集
- CRISPR-mediated gene regulation
- CRISPR導入法
- gRNAデータベース
製品内容 プライスマッチ
カスタムCRISPRベクター
CRISPRゲノム編集は標的細胞内でCas9と標的配列に特異的なgRNAが同時発現している必要があります。当社の直感的なオンラインベクターデザインプラットホームを利用して、30以上のベクターバックボーン(非ウイルス、ウイルス、トランスポゾン)と無数の組み合わせをもつベクターコンポーネント(プロモーター、Cas9バリアント、蛍光タンパク質、薬剤選択マーカーなど)を選択することによって、様々な方法でCas9と任意のgRNAを発現させることができます。さらに、標準プラスミド、レンチウイルス、AAV、アデノウイルス、piggyBacの各ベクターシステムに対して、Cas9とgRNA共発現用のAll-in-oneシステムとデュアルベクターシステムを用意しています。
当社の包括的なCRISPRベクターコレクションには、HDRによる標的サイトへの点変異などの配列改変や大きな配列のノックインをおこなう際にDNAテンプレートとして機能する遺伝子ターゲティング用ドナーベクターも含まれています。さらに、当社は内在性の遺伝子座で標的遺伝子の転写活性化もしくは転写抑制を行う場合に使用する、CRISPRa転写活性化、CRISPRi転写抑制用ベクターも用意しています。CRISPRa用にdCas9-SAMまたはdCas9-SunTagが組み込まれたベクターの設計と作製、CRISPRi用にdCas9-KRABまたはdCas9-KRAB-MeCP2が組み込まれたベクターの設計と作製が可能です。
CRISPRベクターのご注文の際にプラスミドDNA精製サービス、RNA精製サービス、ウイルスパッケージングサービスも追加できます。
設計ヒント
備考: AAVベクターの組み込み可能DNAサイズは4.7kbまでとなります。ポピュラーなStreptococcus pyogenes由来のSpCas9のサイズは4.2kbとなり、プロモーター、ポリアデニレーションシグナル、gRNA発現カセットなどを組み込むならば、AAVの組み込み可能サイズ内に収めることはとても難しくなります。そのため、当社のAAV CRISPRシステムはStaphylococcus aureus由来のサイズの小さい(3.2kb)SaCas9を使用しています。ポピュラーなSpCas9のPAM配列はNGGですが、SaCas9のPAM配列はNNGRRもしくはNNGRRT(推奨)であることにご注意ください。ご希望ならば、SpCas9が組み込まれたAAV CRISPRベクターの作製も請け負っています。
カスタムベクターの選択 表示する
既製CRISPRベクター
カスタムCRISPR/Cas9ベクター作製に加えて、ベクタービルダー社では既製のCas9ベクター群、コントロール用gRNAベクター、CRISPRa及びCRISPRi用ヘルパーベクターを提供しています。これらの既製ベクターは大腸菌ストックの形状でいつでも発送可能です。プラスミドDNA精製サービス、RNA精製サービス、ウイルスパッケージングサービスも既製ベクターのご注文の際に追加できます。
当社の既製CRISPRベクターのコレクションをご覧ください表示する
| ベクターシステム | ベクター名 | ベクターID |
|---|---|---|
| hCas9発現ベクター (標準プラスミド) | pRP[Exp]- mCherry/Hygro-CBh>hCas9 |
VB010000-9378bvk |
| hCas9発現ベクター (レンチウイルス) | pLV[Exp]- CBh>hCas9/Hygro |
VB010000-9380sne |
| SaCas9発現ベクター (AAV) | pAAV[Exp]-CMV>SaCas9 | VB010000-9382per |
| hCas9発現ベクター (アデノウイルス) | pAV[Exp]-CBh>hCas9 | VB010000-9381pwj |
| hCas9発現ベクター (piggyBac) | pPB[Exp]-mCherry/Hygro-CBh>hCas9 | VB010000-9379gqq |
| Scramble gRNAコントロールベクター (標準プラスミド) | pRP[gRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9358ttk |
| Scramble gRNAコントロールベクター (レンチウイルス) | pLV[gRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9359hhe |
| Scramble SagRNAコントロールベクター (AAV) | pAAV[SagRNA]-EGFP-U6>Scramble_SagRNA1 | VB010000-9361zjr |
| Scramble gRNAコントロールベクター (アデノウイルス) | pAV[gRNA]-EGFP-U6>Scramble_gRNA1 | VB010000-9360nph |
| Scramble gRNAコントロールベクター (piggyBac) | pPB[gRNA]-EGFP/Puro-U6>Scramble_gRNA1 | VB010000-9362zuk |
| hCas9 and scramble gRNA共発現ベクター (標準プラスミド) | pRP[CRISPR]-EGFP/Puro-hCas9-U6>Scramble_gRNA1 | VB010000-9354ztt |
| hCas9 and scramble gRNA共発現ベクター (レンチウイルス) | pLV[CRISPR]-hCas9/Puro-U6>Scramble_gRNA1 | VB010000-9355sqw |
| SaCas9 and scramble SagRNA共発現ベクター (AAV) | pAAV[CRISPR]-SaCas9-U6>Scramble_SagRNA | VB010000-9357zmm |
| hCas9 and scramble gRNA共発現ベクター (アデノウイルス) | pAV[CRISPR]-hCas9/EGFP-U6>Scramble_gRNA1 | VB010000-9356pna |
| dCas9-SAM activator MS2/P65/HSF1発現ベクター (レンチウイルス) | pLV[Exp]-EF1A>MS2/P65/HSF1/Hygro | VB010000-9383ffr |
| dCas9-SAM activator dCas9/VP64発現ベクター (レンチウイルス) | pLV[Exp]-EF1A>dCas9/VP64/Bsd | VB010000-9384wwc |
| dCas9-SAM scramble msgRNAコントロールベクター (レンチウイルス) | pLV[msgRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9363gsm |
| dCas9-KRAB-MeCP2発現ベクター (レンチウイルス) | pLV[Exp]-CBh>dCas9/ KRAB/MeCP2/Hygro |
VB010000-9386mwf |
CRISPRウイルス
レンチウイルス、AAV、アデノウイルスは哺乳類細胞にCRISPRコンポーネントを導入するさい、頻繁に利用されます。ベクタービルダー社はトランスフェクションが難しい細胞に対しても高効率CRISPRターゲティングを実現可能にする、プレミアム品質のレンチウイルス、AAV、アデノウイルスのパッケージングサービスを提供しています。当社のウイルスパッケージングは独自の技術や試薬によって大幅に改良されているので、タイター、純度、生存率、再現性の面で優れた結果をもたらします。そのため、当社のクローニング及びウイルスパッケージングサービスに満足され、繰り返しご注文をいただいているお客様の数は増え続けています。
レンチウイルスパッケージングの価格、作業日数、スケールについて表示する
| スケール | 用途 | 通常タイター | 最少タイター |
容量 | 価格(税別、送料別) | 作業日数 |
|---|---|---|---|---|---|---|
| パイロット | 培養細胞 | >4x108 TU/ml | >108 TU/ml | 250 ul (10x25 ul) | 70,000円 | 6-12 日 |
| 中容量 | >3x108 TU/ml | 1 ml (10x100 ul) | 101,000円 | |||
| 大容量 | >2x109 TU/ml | >109 TU/ml | 1 ml (10x100 ul) | 170,500円 | ||
| 超純粋中容量 | 培養細胞、in vivo | >2x109 TU/ml | >109 TU/ml | 500 ul (10x50 ul) | 217,000円 | |
| 超純粋大容量 | 1 ml (10x 100 ul) | 263,500円 |
TU = 形質導入ユニット;Transduction units (または感染ユニット; infectious units)
AAVパッケージングの価格、作業日数、スケールについて 表示する
| スケール | 用途 | 通常タイター |
最少タイター |
容量 | 価格(税別、送料別) | 作業日数 |
|---|---|---|---|---|---|---|
| パイロット | 培養細胞 | >1012 GC/ml | >2x1011 GC/ml | 250 ul (10x25 ul) | 70,000円 | 6-12 日 |
| 中容量 | 1 ml (10x100 ul) | 101,000円 | ||||
| 大容量 | >5x1012 GC/ml | >2x1012 GC/ml | 1 ml (10x100 ul) | 170,500円 | ||
| 超純粋パイロット | 培養細胞、in vivo | >2x1013 GC/ml | >1013 GC/ml | 100 ul (4x25 ul) | 217,000円 | 7-14 日 |
| 超純粋中容量 | 500 ul (10x50 ul) | 310,000円 | ||||
| 超純粋大容量 | 1 ml (10x100 ul) | 480,500円 |
GC = ゲノムコピー
アデノウイルスパッケージングの価格、作業日数、スケールについて表示する
| スケール | 用途 | 通常タイター |
最少タイター |
容量 | 価格(税別、送料別) | 作業日数 |
|---|---|---|---|---|---|---|
| パイロット | 培養細胞 | >2x1010 IFU/ml | >1010 IFU/ml | 250 ul (10x25 ul) | 101,000円 | 28-35 日 |
| 中容量 | 1 ml (10x100 ul) | 170,500円 | ||||
| 大容量 | >2x1011 IFU/ml | >1011 IFU/ml | 1 ml (10x100 ul) | 263,500円 | ||
| 超純粋中容量 | 培養細胞、in vivo | >2x1012 VP/ml | >1012 VP/ml | 500 ul (10x50 ul) | 325,500円 | |
| 超純粋大容量 | 1 ml (10x100 ul) | 387,500円 |
IFU = 感染タイター(Infectious units); VP =ウイルス粒子数 (Virus particles)
Cas9 mRNAとgRNA用のRNA調製
ベクタービルダー社では哺乳類細胞にCRISPRコンポーネントのRNAを導入したい場合に、トランスフェクション対応、マイクロインジェクション対応Cas9 mRNAおよび任意の標的サイトに対するgRNA作製サービスを用意しています。Cas9 mRNAとしてはhCas9ヌクレアーゼとCas9ニッケース(Cas9(D10A)) mRNAが利用可能です。当社ではCRISPRライブラリーデザイン(CLD)のアルゴリズムを利用して計算した特異性スコアといくつかの経験的なルールを使用して、任意の遺伝子/配列をターゲットとする最適なgRNAを設計します。
CRISPR RNA製品の価格と作業日数 表示する
| 試薬 | 濃度と容量 | 価格 (税別、送料別) | 作業日数 |
|---|---|---|---|
| hCas9 mRNA | >500 ng/ul, 25 ul, ヌクレアーゼフリーウォーター, 無菌 | 70,000円 | 2-4 日 |
| Cas9(D10A) mRNA | |||
| Custom gRNA* | 54,500円 |
*gRNAをin virto転写ベクターへクローニングする場合は、23,500円の追加費用と5-10日の作業日数が必要になります。
ゲノム編集用ドナーDNA
ベクタービルダー社ではHDRを利用した切断箇所でのDNA配列編集をおこなうためのドナーDNAテンプレートを、ssODNまたは線状化プラスミドからdsDNAとして作製します。点変異導入やDNA配列のノックインなどに利用できます。ssODNは標的配列への小さいタグや制限酵素サイトなどの短DNA配列(60bp以下)の挿入に利用されます。dsDNAは蛍光タンパク質タグや他のレポーターなどの大きな配列(4-5kbまで)のノックインを行うために利用されます。
ドナーDNAの価格と作業日数表示する
| 試薬 |
価格 (税別、送料別) |
作業日数 |
|---|---|---|
| ssODN (通常 120-200 nt) | 56,000円から | 2-3 週 |
CRISPRライブラリー
ベクタービルダー社では様々なCRISPRライブラリー、例えば、遺伝子の機能スクリーニング用のノックアウトライブラリー、非翻訳領域の遺伝子制御機能スクリーニングや遺伝子の機能獲得型スクリーニング用のCRISPRaライブラリー、遺伝子の機能欠失型スクリーニング用のCRISPRiライブラリーなどのカスタム設計及び構築サービスに特化しています。加えて、1細胞スクリーニング用のCRISPRバーコードライブラリーやその他のCRISPRライブラリーの構築にも対応しています。
構築したライブラリーは大腸菌ストック、プラスミドDNAプール、もしくはウイルスにパッケージングされた形状での納品を選択できます。当社のカスタムライブラリーは次世代シークエンスによって検証され、納品されたライブラリーの検証データの確認も可能です。
カスタムライブラリー構築サービスに加えて、ヒトとマウスの全ゲノムノックアウトスクリーニング用の既製デュアルgRNAレンチウイルスライブラリーも提供しています。デュアルgRNAライブラリーはgRNAペアによって標的遺伝子に大きな配列欠失を導入可能であり、シングルgRNAライブラリーよりも効率的に遺伝子機能欠損変異体を作製できるため、デュアルgRNAライブラリーはシングルgRNAライブラリーよりも強力なノックアウトスクリーニングツールです。ヒトとマウスのデュアルgRNAライブラリーは20048ヒト遺伝子と20493マウス遺伝子を標的とした、即時使用可能な高タイターのレンチウイルスの形状で構築されています。各遺伝子は可能な限り4-6種の異なるgRNAペアによってターゲットとなるように設計されています。
既製デュアルgRNA製品の価格と作業日数表示する
| 製品名 | 遺伝子数 | gRNAペア数 | スケール | カタログ番号 | 価格(税別、送料別) |
|---|---|---|---|---|---|
| ヒト全ゲノムデュアルgRNA レンチウイルスライブラリー | 20,048 | 91,926 | 中容量 (>1.0x108 TU/ml, 1 ml) |
LVM(Lib190505-1046fgb) |
384,000円
|
| プラス (>1.0x108 TU/ml, 5 ml) |
LVM(Lib190505-1046fgb) |
816,000円
|
|||
| マウス全ゲノムデュアルgRNA レンチウイルスライブラリー | 20,493 | 90,344 | 中容量 (>1.0x108 TU/ml, 1 ml) |
LVM(Lib190505-1050kpm) |
384,000円
|
| プラス (>1.0x108 TU/ml, 5 ml) |
LVM(Lib190505-1050kpm) | 816,000円 |
CRISPRソリューションズ
CRISPRシステムの高い汎用性によって哺乳類細胞での様々なゲノム編集が可能になりました。当社の分子生物学に精通した技術陣があらゆるCRISPRゲノム編集プロジェクトにおいて、実験設計から試薬の生産までの完全なサポートを行います。以下に記載した一般的なCRISPRプロジェクトのサポート、または新規のCRISPRプロジェクトに協力します。デザインサポートを依頼する に詳細を送っていただければ、当社からのサービス内容を無料で提案させていただきます。
- 点変異の導入
-
DNA配列のノックイン
- 小さいタグ配列の挿入 (< 60 bp)
- 大きなDNA配列の挿入 (4-5 kbまで)
- CRISPR遺伝子転写活性化 (CRISPRa)
- CRISPR遺伝子転写不活性化 (CRISPRi)
- CRISPRライブラリー (ノックアウト, CRISPRa, CRISPRi, バーコードなど)
-
安定細胞株作製
- Cas9発現細胞株
- iPSCゲノム編集
- がん細胞ゲノム編集
技術情報
CRISPRゲノム編集
従来のCRISPRシステムは2つのコンポーネント(Cas9とガイドRNA(gRNA))から構成されます。もっとも広く利用されているCas9はStreptococcus pyogenesをもとに開発され、SpCas9またはhCas9と呼ばれるRNA誘導性DNAヌクレアーゼであり、標的とするDNA配列に2本鎖切断(DSB)を作り出します(図1)。他のCas9バリアントとしてCas9ニッケース(Cas9(D10A))があり、DNA鎖上に一本鎖切断を作り出します。Cas9ニッケースが標的配列のおける相補鎖のぞれぞれの鎖を標的とする2つのgRNAとともに使用されると、ニッケースは各鎖に対し1か所ずつ、2か所の一本鎖切断を標的配列に作り出すことによってDSBが生じます。CRISPRによるDSB形成後、細胞は非相同末端結合(NHEJ)パスウェイを活性化させてDNA損傷を修復するが、その際に配列の小規模な欠失、挿入及び配列変換が起こります。それらの変異が遺伝子の翻訳領域で発生すると、遺伝子機能の破壊につながります。

図1. CRISPRによるDNA切断後のDNA修復機構
また、外来性のドナーDNAテンプレートが存在する場合には、効率はやや低いもののDSBが相同組み換え修復(HDR)によって修復されることもあります。HDRによって標的ゲノムDNA配列は外来性テンプレートDNAに置き換えられ、標的配列においてドナーDNAのノックインや点変異などの配列改変が起こります。ドナーDNAテンプレートとして一本鎖オリゴヌクレオチド(ssODN)もしくはdsDNA(線状化したプラスミドDNA)が使用されます。ssODNは点変異や小さいタグ配列の挿入に適し、dsDNAは大きなDNA配列をノックインする用途に適しています。

図 2. gRNA-Cas9リボ核タンパク質 (RNP) を使用して、ホモ接合型 CD274ノックアウト (KO) 変異体を作成した。 (A) 編集されたRNP は、標的細胞にエレクトロポレーションで導入し、単一クローンがスクリーニングによって分離された。候補細胞の遺伝子型は、PCR およびサンガーシーケンスによって検証した。(B) マウス結腸腺癌細胞株を編集するこのケーススタディでは、標的遺伝子の2か所に結合するRNPを使い、13kbp 領域をKOするべく細胞にエレクトロポレーションした。 P1からP4の4種類のプライマーをデザインし、3か所のPCRフラグメント長を比較することで、KOクローンとWTクローンを判別した。(C) PCR の結果に基づいて、クローン1はホモ接合型KO変異体と判断された。(D) 配列を確認した。
CRISPRを使った遺伝子制御
不活性型Cas9 (dCas9)を他の転写調節補助因子と組み合わせることで、内在ゲノムの遺伝子座の転写制御が可能になります。内在性遺伝子の転写活性には、相乗的活性化因子(Synergistic Activation Mediator : SAM)システムをが利用できます。このシステムでは、次の3種類のコンポーネントを用います: dCas9/VP64融合タンパク質, MS2/P65/HSF1ヘルパー、そして改変(modified) gRNAです。dCas9タンパク質は、合成転写活性化因子VP64と組換え融合タンパク質を生合成します。gRNAはMS2アプタマーシークエンスもフランキングにデザインして改変しています。そのため、NF-kB (p65)およびHSF1のトランスアクティベータードメインからなるMS2複合体をリクルートします。VP64は、HSV(ヘルペス単純ウイルス)タンパク質16の4回タンデムリピートから構成され、Cas9タンパク質のC末端に結合することで、転写活性化マシーナリーをリクルートします。NF-kB (p65) およびHSF1のトランス活性化ドメインは転写活性化因子として働き、さらに転写活性化因子のコファクターをプロモーターやエンハンサーにリクルートします。よって、dCas9/VP64とMS2/P65/HSF1複合体は図3に示すように共同的に遺伝子発現を活性化します。 SAMシステム以外にもVectorBuilderではdCas9-VP64-p65-Rta (VPR) トランスアクティベーションドメインシステムも提供しています。このシステムでは、HSF1のトランスアクティベーションドメインの代わりに、Epstein-Barr ウイルスRトランスアクティベーターが用いられています。
一方、dCas9をKruppel-associated box domain (KRAB) や MeCP2といった転写抑制因子と組み合わせることで遺伝子発現の抑制が可能です。VectorBuilderでは、現在、2種類のdCas9/KRAB ヘルパーベクターを提供しています。一方は従来のdCas9/KRABヘルパーベクターと、他方はdCas9とKRAB/MeCP2の2つの抑制ドメインを融合させ、標的DNAの転写抑制効果を向上させた改良型です。図4に示すように、gRNAがdCas9/KRAB/MeCP2をゲノム中の内在プロモーターにリクルートし、ターゲット遺伝子の転写が抑制されます。KRABドメインはヒトの転写因子によくみられる構造で、ヒトゲノム中で同定されている最も転写抑制効果の高い因子の一つです。MeCP2はヒストン脱アセチル化酵素をリクルートすることで、エピジェネティック修飾とクロマチン凝縮を引き起こし、KRABによる転写抑制効果を高めます。CRISPRを使った遺伝子制御では、転写制御因子のクロマチンへの影響の研究、目的遺伝子の発現制御、全ゲノムスクリーニングなど、様々な活用法があります。
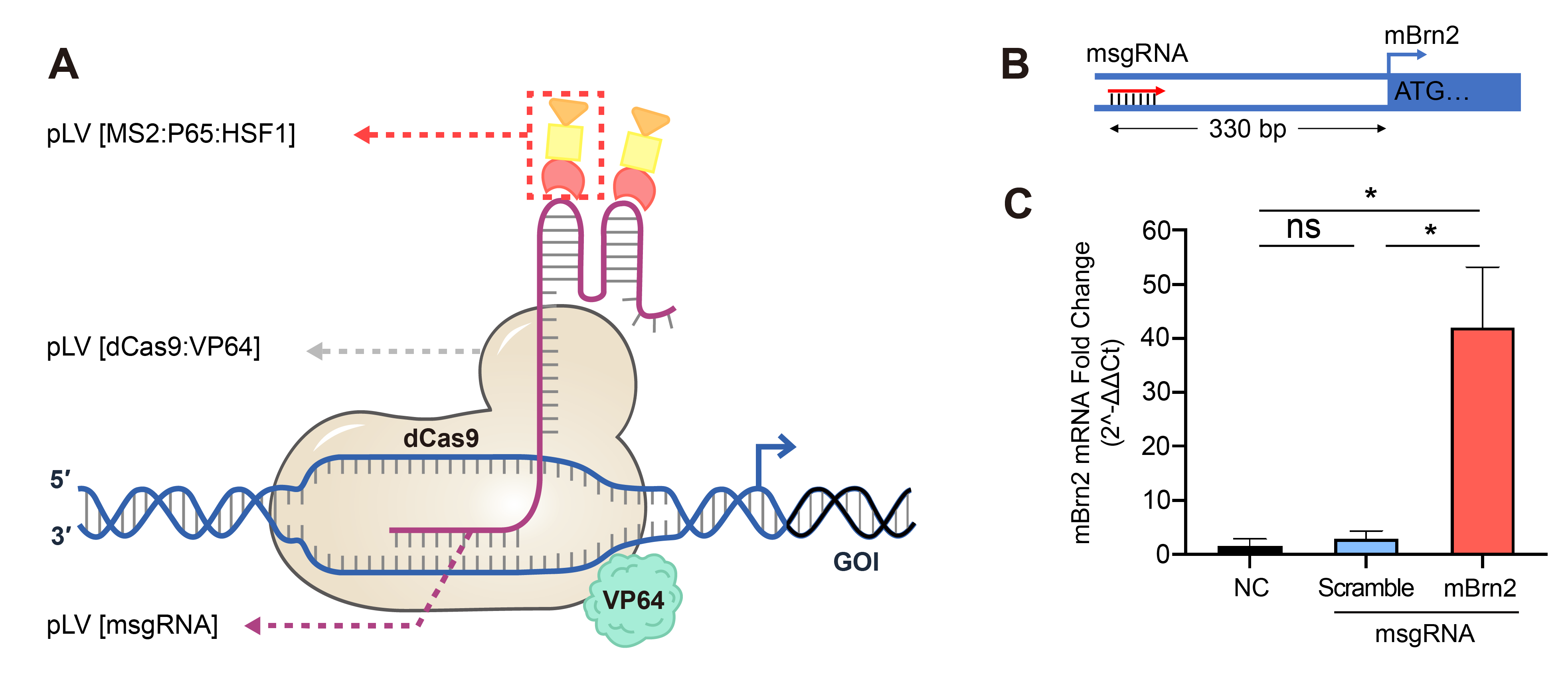
図 3. レンチウイルスを用いたCRISPRa による遺伝子発現の活性化。SAM複合体, dCas9/VP64, および MS2/P65/HSF1を安定発現するNIH3T3細胞に、msgRNA 発現レンチウイルスを感染させ、抗生物質選択を行った。 (A)SAMシステムによる転写活性制御の図解。 (B) マウスBm2遺伝子のプロモーター領域を標的としたmsgRNA の図。(C) NIH3T3細胞にスクランブルまたはターゲッティングmsgRNA を導入したもの、または無処理コントロール (NC)のBm2遺伝子発現量をqRT-PCR法にて比較測定した結果。 Mean±SD, *P<0.05, Tukey法によるANOVA。
.png")
図 4. レンチウイルスを用いたCRISPRiによる遺伝子発現抑制。dCas9/KRAB/MeCP2転写抑制複合体を安定発現するJurkat細胞にgRNA 発現レンチウイルスを感染させ、抗生物質選択を行った。(A) dCas9/KRAB/MeCP2による遺伝子転写抑制の図解。 (B) ヒトCXCR4遺伝子(ケモカインレセプター)のプロモーター領域を標的としたgRNA設計の図。(C)Jurkat 細胞にスクランブルまたはターゲッティングgRNA を導入したもの、または無処理コントロール (NC)のCXCR4遺伝子発現量をqRT-PCR法にて比較測定した結果。Mean±SD, ***P<0.001, ****P<0.0001, Tukey法によるANOVA。(D)Jurkat 細胞にスクランブルまたはターゲッティングgRNA を導入したもの、または無処理コントロール (NC) におけるCXCR4タンパク質発現量をウェスタンブロット法によって比較した結果を示す。(E)スクランブルまたはターゲットgRNAで形質導入されたJurkat細胞膜に発現するCXCR4をフローサイトメトリーで定量化した。CXCR4は、モノクローナル一次抗体 (Ab) および蛍光色素結合型二次抗体で標識した。ネガティブコントロールには、無標識二次抗体と、Jarkat細胞を用いた。 (F) CXCR4をターゲットするgRNAで形質導入された細胞の表面上のCXCR4タンパク質量は、スクランブルgRNAを形質導入された細胞に比較して、平均して約50%減少した。Mean±SD。
CRISPR導入法
CRISPRコンポーネントの哺乳類細胞への導入方法は複数あり、以下に示します(図5)。
- gRNA、Cas9プラスミド
- gRNA、Cas9 ウイルス (レンチウイルス, AAV, アデノウイルスなど)
- gRNA、Cas9 mRNA混合物
- gRNA-Cas9タンパク質複合体(RNP)形成
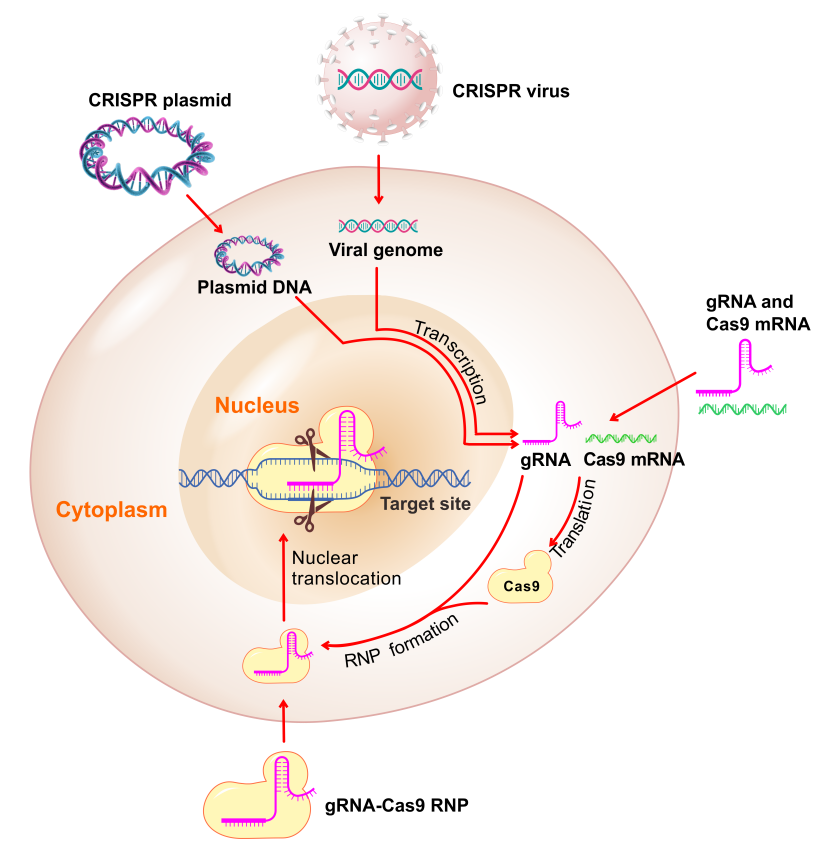
図5. 細胞へのCRISPRコンポーネントの導入方法
下表にそれぞれの導入方法の利点と欠点を記載しました。実験内容に応じて最も適した導入方法を選択してください。
| 導入方法 | 利点 | 欠点 |
|---|---|---|
| gRNA、Cas9プラスミド |
|
|
| gRNA、Cas9ウイルス |
|
|
| gRNA、Cas9 mRNA混合物 |
|
|
| gRNA-Cas9タンパク質複合体(RNP)形成 |
|
|
gRNAデータベース
ベクタービルダー社のオンラインベクター設計ツールは最適化されたヒト、マウス、ラットの全ゲノムデータベースを提供しており、高効率なターゲティング効率をもつCRISPRベクターの設計を可能にします。gRNA特異性スコアの計算はCRISPRライブラリーデザイン(CLD)のアルゴリズムを使用しています。 簡単に説明すると、ある生物種のN(20)NGG配列を標的とするgRNAを設計する場合、当社はゲノム上で標的配列から3つまでのミスマッチを含む、潜在的なオフターゲット配列すべてを検索します。各潜在的オフターゲット配列からオフターゲットスコアを計算したのち、すべてのオフターゲットスコアを併せて、最終的なgRNAスコア (0-100:高いスコアは高い特異性を意味する) を算出します。gRNAスコアは大まかな指標に過ぎないことに留意してください。実際のターゲティング効率と特異性はスコアとは異なる可能性があります。gRNAスコアが低くても十分に機能する場合もあります。
ベクタービルダー社のオンラインベクター設計ツールでCRISPRベクターを設計する場合、当社のデータベースで標的遺伝子を探すこともできます。遺伝子名を入力していただければ、当社データベースで標的遺伝子をターゲットとして設計された、すべてのgRNAの詳細な情報を閲覧できます。当社の全ゲノムgRNAデータベースを使っていただければ、別のgRNA設計ツールやソフトウェアを必要としないで、標的配列の検索/解析をしなくても任意の遺伝子に対する適切なgRNA配列を得ることができます。
ベクタービルダー社HPの“リソース”>“ラーニングセンター”の豊富な教育リソースによって、お客様のCRISPR実験の計画、遂行、トラブルシューティングをサポートしています。
CRISPRベクターシステムの概要はこちらからCRISPRコンポーネントの概要はこちらから
リソース
技術関連資料
Q&A
目的遺伝子の働きを不活性化し、その影響を調べる目的ではshRNAノックダウンもCRISPRやTALENを使ったノックダウンのどちらも役に立ちます。あなたの目的にとってどちらがより適しているかを判断するためのポイントを以下に記載します。
メカニズム
ノックダウンベクター
ノックダウンベクターはshRNAという短いヘアピン状のRNAによって細胞の標的mRNAを分解、翻訳阻害することで目的遺伝子の機能を抑えます。そのため、shRNAノックダウンベクターは標的遺伝子配列を改変することはありません。
ノックアウトベクター
CRISPRとTALENはどちらもヌクレアーゼにゲノム上の標的配列を切断するように働きかけます。分解された配列が不完全な形で修復され結果、配列の挿入もしくは欠損が起こり、恒久的な変異となります。それらの変異のいくつかは翻訳コドンのずれ(フレームシフト)やストップコドンを作り出し、標的遺伝子の機能不全を引き起こします。ゲノム上で数kb以内にふたつの標的配列が同時に切断された場合は、その間の配列に欠損が起こることもあります。
効率
shRNAノックダウンは最も効果的なshRNAをつかったとしても、標的遺伝子の発現を100%抑制することはできません。一方でCRISPRやTALENの場合、一部の細胞で遺伝子の機能を完全に欠損させる変異を持つ細胞を作成できます。
再現性と均一性
一般的にshRNAベクターは、細胞プール内のほとんどの細胞から均一な結果が得られ、複数の実験の間で高い再現性が得られます。一方でCRISPRやTALENは変異が偶発的に引き起こされるので、得られる結果が細胞によって違います。細胞の標的遺伝子を完全に欠損させるためには細胞内のすべての遺伝子コピーを破壊しなくてはなりません。X-またはY-染色体上の遺伝子を除いて大抵の細胞は遺伝子が2コピーあり、そしてがん細胞は2コピー以上の遺伝子を持つので遺伝子を完全に欠損させた細胞が得られる可能性はとても低くなります。このような理由から、CRISPRやTALENのようなヌクレアーゼを利用した遺伝子ノックアウトはすべての標的遺伝子コピーが破壊された細胞をDNAシークエンスによって特定する必要があります。
オフターゲット効果
オフターゲット効果はshRNAノックダウンでもCRISPRやTALENノックアウトでも報告されています。観察された表現型がオフターゲット効果であるかどうかは同じ標的遺伝子に対して異なる複数のshRNAを使うことで確認できます。もし、異なる複数のshRNAノックダウンから得られた表現型が同じならば、その表現型はオフターゲット効果ではない証拠となります。CRISPRやTALENノックアウトを使う場合は、オフターゲット効果による表現型でないことを確認するために標的遺伝子に対して異なる機能欠損変異を持つ複数のクローンを作成して解析する必要があります。加えて、得られた変異体クローンのゲノムから、バイオインフォマティクスによって推測されるオフターゲットとなりうる配列をシークエンスして、それらに変異がないことを確認する方法もあります。
CRISPRとTALENは両方とも培養細胞およびモデル生物のゲノム編集用途に開発されました。どちらも遺伝子のノックアウトや点変異、挿入のノックインに使用できますが、いくつか違いがありそれぞれ長所と短所があります。
メカニズム
CRISPR
CRISPRは部位特異的ガイドRNA(gRNA)を利用して、Cas9ヌクレアーゼにゲノム内の標的部位のDNA配列を切断させます。標的部位は通常およそ20bpの長さであり、数塩基の配列の違いがあっても認識されて切断されてしまうことがあります。
TALEN
TALENは、FokIヌクレアーゼドメインに融合したTALエフェクターDNA結合ドメイン(特異的配列を認識する)で構成されるキメラタンパク質を2つで一対のペアとして利用します。タンパク質のペアは、14‐20bp(スペーサー領域)を挟んだ2つの標的部位(∼18bp)に結合するように設計されています。標的部位DNAに結合すると、Foklヌクレアーゼドメインは二量体を形成し、続いてスペーサー領域のDNAを切断します。
効率
CRISPRとTALEN両方とも効率良くゲノム編集できますが、用途、動物種、細胞タイプによって効率は大きく変わります。一般に、CRISPRはTALENよりも効率的に細胞内に導入され、DNAを切断します。
オフターゲット効果
CRISPR gRNAは∼20 bpの配列を標的とし、一方TALENペアは、合計∼36bpの標的配列に結合します。Cas9/gRNA複合体はTALENよりも標的配列とミスマッチがある配列(最大で5bpまで)に結合する可能性があります。したがって、TALENによる切断はCRISPRよりもオフターゲット切断の可能性は低く、高い特異性があります。CRISPRを培養細胞株につかうとオフターゲット効果がみられますが、CRISPRノックアウトマウスの分析からCRISPRを生体につかった場合(in vivo)、オフターゲット効果の頻度は低くなることが報告されています。近年のCRISPRシステムの改良によりCRISPRの特異性は大幅に上昇しました。変異触媒ヌクレアーゼドメイン(例えばCas9_D10AもしくはCas9_H840A)を含むCas9ニッカーゼをデュアルgRNAと一緒に使用することで標的領域の内で2つの一本鎖DNAニックが生成され、ダブルニックDSB(二重鎖破壊)ができます。このデザインでは2つのgRNAを使うことで標的配列が∼40bpに拡張され、オフターゲット効果が起こる可能性を最小限にできます。
標的配列の必要条件
一方、CRISPRはgRNA標的配列の3'末端にPAM配列(通常NGG)が必要となります。遺伝子のノックアウトのみが目的ならばCRISPRを使っても支障はありませんが、遺伝子の特定の部位での切断を必要とする部位特異的突然変異または挿入を導入するときに問題があります。CRISPRを使って特定のゲノム部位に正確な編集をするならば、相同組換えドナーベクターまたは編集標的配列の近傍配列を含むオリゴヌクレオチドをgRNAとCRISPRを共に細胞に導入することで、HDR(相同性組み換え修復)を引き起こして標的部位を正確に編集する方法があります。
簡便性
シンプルさでは、CRISPRはいくつかの点でTALENより優れています。第一に、ベクター構築の時に、CRISPRのCas9/gRNA複合体の標的への結合は単純なRNA/DNAハイブリダイゼーションに依存するため、短いgRNAをデザインするだけですが、TALENはタンパク質とDNAの相互作用ごとに特別なTAL DNA結合ドメインの再エンジニアリングを必要とします。そのため、gRNAの設計は、標的配列毎に常に 2つのベクターを必要とするTALENよりも安くて簡単です。しかし、ベクタービルダー社が用意したTALEN認識モジュールはTALENベクターの設計作業を大幅に削減できます。第二に、マウス胚への注入などの一部のアプリケーションにおいてCas9タンパク質およびgRNAの直接注射による効率的な導入が可能ですが、TALENでは同様のことをできません。第三に、CRISPRは、数千の異なるgRNAを発現するCRISPRライブラリーを作成し、ハイスループットでスクリーニングすることが容易であるため、汎用性の高い遺伝子スクリーニングが可能です。
CRISPRを利用したゲノム編集では、Cas9ヌクレアーゼはゲノム上のgRNAの標的配列に誘導され、DNAを切断します。単純な遺伝子破壊株を作成する場合、大抵はシングルgRNAとCas9をつかってDNA二本鎖切断(double-strand break 、DSB)と非相同性末端再結合(non-homologous end joining、NHEJ)を起こして標的配列に小さな挿入/欠損等の変異を導入します。それらの変異のいくつかはフレームシフトや停止コドンを創り出し、遺伝子機能を破壊します。
デュアルgRNAはCas9(D10A)ニッケースと共につかうと標的配列の2本鎖DNAのそれぞれの鎖を標的とします。ニッケースがデュアルgRNAのそれぞれに誘導されてDNA鎖の片方一本ずつを切断し、二本鎖切断を作り出します。この手法では二つのgRNAが二本鎖切断に必要になるのでCRISPR/Cas9によるオフターゲット効果を減らすことができます。
デュアルgRNAはCas9(D10A)ニッケースと外来性ドナーDNAを使うことで、目的遺伝子に任意の塩基置換(ノックインなど)を導入することも可能です。変異を導入したい箇所を挟んだ近接配列の一つずつを標的とする二つのgRNAによって切断箇所の修復に伴った外来性ドナーDNAをつかった相同組み換え修復(homology-directed repair、HDR)が起こります。