



shRNA遺伝子KDソリューションズ
VectorBuilderでは、RNAi実験に対応した様々なshRNA関連ツールをご提供しています。U6およびmiR30ベースの両システムをご用意していますので、実験のニーズに合わせて自由にカスタマイズいただくことにより、フレキシブルにshRNA発現を制御することができます。ウイルスパッケージングサービス(レンチウイルス、AAV、アデノウイルスなど)もご提供しておりますので、トランスフェクションが難しい細胞には、ウイルスを使用してshRNAツールを導入することも可能です。また大規模な機能喪失スクリーニングには、プール型shRNAライブラリーの構築サービスもご用意しています。よく使われるモデル生物のshRNAデータベースとオンラインベクターデザインプラットフォームを使用すれば、目的遺伝子(GOI)に最適なshRNAを簡単に見つけ、ベクターデザインができます。
shRNAの特長
- 全ゲノムshRNAデータベースが、ベクタービルダーオンラインのベクターデザインポータルに統合されているため、ベクターデザインとshRNAの選択が簡単に行える。
- ベクターバックボーンとベクターコンポーネントの豊富なコレクション。
- 既製品shRNAライブラリーとカスタムメイドshRNAライブラリーに対応。
- 100% シークエンス保証。他社が追随できないお得価格と早い作業日数で受託サービスをお届け。
- shRNAの選択やベクターデザイン、トラブルシューティングなどきめ細やかな技術サポート をご提供。
受託サービスの種類
- カスタムshRNAベクター
- ポピュラーshRNAベクター
- shRNAウイルス
- shRNA (3+1)ウイルスパッケージング
- プール型shRNAライブラリー
- shRNAノックダウン安定細胞株
技術的情報
- shRNAを介した遺伝子ノックダウン
- shRNAの発現コントロール
- 実験による検証
- shRNAデータベース
shRNA受託サービスの種類 プライスマッチ
カスタムshRNAベクター
直観的に使えるオンラインデザインプラットフォームには、30種類以上のベクターバックボーン(非ウイルス用、ウイルス用、トランスポゾン用) と、無制限の組みあわせが可能なベクターコンポーネント (プロモーター、蛍光マーカー、薬剤耐性マーカーなど) が自由自在に選択できるようになっているため、希望通りのベクターデザインにカスタマイズすることができます。ベクタービルダーでは、shRNA データベースがベクターデザインポータルに直結しているため、目的遺伝子に対して、最適なshRNAを簡単に選択することができます。ノックダウン効率のテストには、 shRNAセンサーベクターもご用意していますのでお試しください。
プラスミドDNA精製や、ウイルスパッケージングは関連サービスとして、ベクター構築サービスに追加して同時注文を行っていただけます。
カスタムshRNAベクターをデザインする表示する
既存ベクターシステムに加えて、IPTG誘導性 shRNA 発現ベクターや、 Cre-loxのコンディショナルshRNA発現ベクターもデザインしてベクター構築の受託作製をお受けしています。 デザインサポートを依頼する からお問合せください。
価格は予告なく変更される場合があります。
ポピュラーshRNAベクター
VectorBuilderでは、コントロールベクターとしてスクランブルshRNAや、盛んに研究されているポピュラー遺伝子をターゲットとしたshRNAベクターをご用意しています。すでにデザインされているベクターをショッピングカートに追加していただければご注文も簡単です。プラスミドDNA精製や、ウイルスパッケージングは関連サービスとして、ベクター構築サービスに追加して同時注文を行っていただけます。
U6ベースのポピュラーshRNAベクターは、下のプルダウンピッカーから簡単選択を行っていただけます。
miR30ベースの ポピュラーshRNAベクターは デザインサポートを依頼する からご希望のベクターデザインをご記入の上、お問合せください。
shRNAウイルス
VectorBuilderでは、ウイルスパッケージングサービス(レンチウイルス、AAV、アデノウイルスなど)もベクター構築に合わせてご提供しています。トランスフェクションが難しい細胞には、ウイルスを使用して目的shRNAのツールを導入することも可能です。パッケージングサイズは、目的に合わせて様々なサイズからお選びいただくことができます。パッケージングは当社独自の技術・試薬で改良されたオリジナルプロトコールで、当社のウイルスベクターシステムに最適化されています。タイター、純度、導入効率、再現性に優れているため、リピーターも多く、満足度の高い弊社サービスの一つとなっています。
レンチウイルスパッケージングサービスの注文情報 表示する
| サイズ | 使用目的 | 標準タイター | 最小タイター | 容量 | 価格(消費税・送料別) | 作業日数 |
|---|---|---|---|---|---|---|
| パイロット | 培養細胞 | >4x108 TU/ml | >108 TU/ml | 250 ul (10x25 ul) | 70,000円 | 6-12 日 |
| 中容量 | >3x108 TU/ml | 1 ml (10x100 ul) | 101,000円 | |||
| 大容量 | >2x109 TU/ml | >109 TU/ml | 1 ml (10x100 ul) | 170,000円 | ||
| 超純粋中容量 |
培養細胞& in vivo |
>2x109 TU/ml | >109 TU/ml | 500 ul (10x50 ul) | 217,000円 | |
| 超純粋大容量 | 1 ml (10x 100 ul) | 263,500円 |
TU = Transduction units(トランスダクションユニット、感染タイターとも呼ばれる)
価格は予告なく変更される場合があります。
AAVパッケージングサービスの注文情報表示する
| サイズ | 推奨使用系 | 標準タイター | 最小タイター | 容量 | 価格(消費税・送料別) | 作業日数 |
|---|---|---|---|---|---|---|
| パイロット | 培養細胞 | >1012 GC/ml | >2x1011 GC/ml | 250 ul (10x25 ul) | 70,000円 | 6-12日 |
| 中容量 | 1 ml (10x100 ul) | 101,000円 | ||||
| 大容量 | >5x1012 GC/ml | >2x1012 GC/ml | 170,500円 | |||
| 超純粋パイロット | 培養細胞 & in vivo | >2x1013 GC/ml | >1013 GC/ml | 100 ul (4x25 ul) | 217,000円 | 7-14日 |
| 超純粋中容量 | 500 ul (10x50 ul) | 310,000円 | ||||
| 超純粋大容量 | 1 ml (10x100 ul) | 480,500円 |
GC = Genome copies(ゲノムコピー数)
価格は予告なく変更される場合があります。
アデノウイルスパッケージングサービスの注文情報表示する
| サイズ | 推奨使用系 | 標準タイター | 最小タイター | 容量 | 価格(消費税・送料別) | 作業日数 |
|---|---|---|---|---|---|---|
| パイロット | 培養細胞 | >2x1010 IFU/ml | >1010 IFU/ml | 250 ul (10x25 ul) | 101,000円 | 28-35 日 |
| 中容量 | 1 ml (10x100 ul) | 170,500円 | ||||
| 大容量 | >2x1011 IFU/ml | >1011 IFU/ml | 1 ml (10x100 ul) | 263,500円 | ||
| 超純粋中容量 | 培養細胞 & in vivo | >2x1012 VP/ml | >1012 VP/ml | 500 ul (10x50 ul) | 325,500円 | |
| 超純粋大容量 | 1 ml (10x100 ul) | 387,500円 |
IFU = Infectious units(感染タイター); VP = Virus particles(ウイルス粒子)
価格は予告なく変更される場合があります。
shRNA (3+1)ウイルスパッケージング
ノックダウン効率はshRNAの長さ、ループ構造、shRNAのGC含量および熱力学的安定性、標的配列の二次構造、さらに他の遺伝子とのオフターゲットマッチなど、複数の様々な要因によって決まってきます。そのためアルゴリズムに基づいてデザインされた高いスコアーのshRNAを使ったとしても、強いノックダウン効果が得られない場合もあります。通常~50-70%のshRNAにはある程度のノックダウン効果が観察されますが、強力なノックダウン効果が観察されるのは、そのうちの~20-30%です。この問題を克服するために、複数のshRNAをテストし、最適なshRNAを使うことが結果への近道です。
VectorBuilderでは shRNA (3+1) ウイルスパッケージングサービスをご提供しています。このサービスでは、目的遺伝子をターゲットする3種類のカスタムshRNAベクターのウイルスパッケージングと、1種類のスクランブルコントロールshRNAレンチウイルスのパッケージングをセットにして、割引価格でご提供しています。このサービスは現在、レンチウイルス、AAV、アデノウイルスパッケージングに対応しています。

shRNA (3+1) ウイルスパッケージングサービスの注文情報表示する
| ウイルス種 | サイズと納品形態 | 推奨使用系 | 価格 (消費税・送料別)* | 作業日数** |
|---|---|---|---|---|
| レンチウイルス | パイロット | 培養細胞 | 232,500円 | 10-19 日 |
| 中容量 | 310,000円 | |||
| 大容量 | 465,000円 | |||
| 超純粋中容量 | 培養細胞 & in vivo | 620,000円 | ||
| 超純粋大容量 | 744,000円 | |||
| AAV | パイロット | 培養細胞 | 232,500円 | 10-19 日 |
| 中容量 | 310,000円 | |||
| 大容量 | 465,000円 | |||
| 超純粋パイロット | 培養細胞 & in vivo | 651,000円 | 11-21 日 | |
| 超純粋中容量 | 883,500円 | |||
| 超純粋大容量 | 1,364,000円 | |||
| アデノウイルス | パイロット | 培養細胞 | 372,000円 | 34-47 日 |
| 中容量 | 558,000円 | |||
| 大容量 | 728,000円 | |||
| 超純粋中容量 | 培養細胞 & in vivo | 961,000円 | 34-47 日 | |
| 超純粋大容量 | 1,162,500円 |
* 価格には、ベクター構築とウイルスパッケージングの全てを含みます。
** 作業日数はベクター構築とウイルスパッケージングの両工程を合計しています。
*** 価格は予告なく変更される場合があります。
ノックダウン保障:shRNA (3+1)ウイルスセットを、当社のshRNAリストからトップ3を選択されたにも関わらず、目的遺伝子のノックダウンが >=70%を達成できなかった場合、4種類目のshRNA Virusを無料でご提供します。(注:4種類目のウイルスをドライアイス発送する際、送料手数料のご負担が発生いたします。ご了承ください)。
下の選択ツールを使って目的遺伝子のshRNA (3+1) ウイルスパッケージングを注文する
注:ベクターピッカーは U6ベースshRNAベクターデザインにのみ対応しています。miR30ベースshRNA ベクターのデザインやカスタム性の高いshRNAベクターのデザインは、 デザインサポートを依頼する からベクターデザインの情報とともにお問合せください。
shRNAライプラリー(プール型)
カスタムライブラリー:疾患機序、薬物治療に対する細胞応答、発生プロセス、遺伝子制御などに関与する遺伝子の大規模な機能欠失(loss-of-function)スクリーニングを行うために、強力かつコストパフォーマンスの高いツールです。ライブラリーは、大腸菌ストック、プラスミドDNAプール、パッケー人ぐ済のレンチウイルスなど、ユーザーのご希望に合わせた形態に対応します。カスタムライブラリーは、次世代シークエンシングで完全に検証後、シークエンス情報とともにお届けします。
既製品プリメイドshRNAライブラリー:ヒトまたはマウスの全ゲノム (~19,000 RefSeq genes) またはエリート遺伝子 (PubMed Centralで、引用数の高い上位~2,000遺伝子)があり、eady-to-useの高タイターのレンチウイルスで納品します。各遺伝子は5-6種の異なるshRNAでターゲットされています。ライブラリーの品質は次世代シークエンシング(NGS)と機能アッセイで完全に検証後出荷いたします。
既製品shRNAライブラリーの特長:
- 全ゲノムへのターゲッティング、かつ高いカバー率のターゲッティング
- NGSによるライブラリー品質の検証
- 高い均質性
- ready-to-useの高タイターのレンチウイルス
- EGFP/Puroの両マーカーを発現させるウイルスのため、導入された細胞の選別が目的に合わせて効果的に行える
プリメイド(既製品)shRNAライブラリーの注文情報表示する
| 製品名 | 遺伝子数 | shRNA数 | スケール | カタログ番号 | 価格 (消費税・送料別) | 作業日数 | |
|---|---|---|---|---|---|---|---|
| ヒト エリート遺伝子 プール型 shRNA ライブラリー | 2,161 | 12,471 |
中容量 (>1.0x108 TU/ml, 1 ml) |
LVM (Lib190505-1037bjk) | 232,500円 | 7-14 日 | |
| マウス エリート遺伝子 プール型 shRNA ライブラリー | 2,233 | 12,472 |
中容量 (>1.0x108 TU/ml, 1 ml) |
LVM (Lib190505-1039sgb) | 232,500円 | ||
| ヒト ホールゲノムプール型shRNA ライブラリー | 20,593 | 105,233 |
中容量 (>1.0x108 TU/ml, 1 ml) |
LVM (Lib230926-1079mym) | 232,500円 | ||
|
プラス (>1.0x108 TU/ml, 5 ml) |
LV5M (Lib230926-1079mym) | 697,500円 | |||||
| マウスホールゲノム プール型shRNA ライブラリー | 22,023 | 105,170 |
中容量 (>1.0x108 TU/ml, 1 ml) |
LVM (Lib230926-1080rpt) | 232,500円 | ||
|
プラス (>1.0x108 TU/ml, 5 ml) |
LV5M (Lib230926-1080rpt) | 697,500円 |
* エリート遺伝子ライブラリー:中容量は 100 x shRNAカバレッジで、 >40スクリーニングを実施できます。ホールゲノムライブラリー:中容量は 100 x shRNAカバレッジで、 >5 スクリーニングを実施できます。プラススケールの場合、 100x shRNAカバレッジで、>25 スクリーニングを実施できます。
** 価格は予告なく変更される場合があります。
shRNAノックダウン安定発現細胞株
VectorBuilderでは、GOIの長期ノックダウンが必要なアプリケーション向けに、shRNA安定細胞株樹立サービスをご提供しています。ノックダウンスコアの上位3位のshRNA候補ターゲットシークエンスをテストし、ノックダウン効率が最も高かったshRNAをレンチウイルスで細胞に導入して安定細胞株を作製します。細胞株のノックダウンレベルはRT-qPCRで検証します。単離された細胞株は、無菌試験やマイコプラズマ検出など一連の標準的なQCアッセイに合格して出荷されます。
安定細胞株受託構築の注文情報表示する
| 安定発現細胞株モデル | 樹立方法 | 納品形態 | 価格 (消費税・送料別) | 作業日数 |
|---|---|---|---|---|
| shRNA遺伝子ノックダウン | レンチウイルスでの遺伝子導入 | 細胞プール (>106 細胞/バイアル,2本) | 62,0000円より | 8-13週間 |
| 2 モノクローン (>106 細胞/バイアル,2本/モノクローン) | 775,000円より | 13-18週間 |
* 価格は予告なく変更される場合があります。
shRNA関連のテクニカルインフォメーション
shRNAを介した遺伝子ノックダウン
ショートヘアピン RNA(shRNA)は、ステムループ構造を持つ RNA 分子であり、相補的な塩基対形成を介して標的mRNAを分解できるため、様々な RNAi アプリケーションに広く利用されています。 shRNAは2本鎖DNAベクターを用いて、トランスフェクションやウイルス感染などの手法で、細胞に導入することができます。 shRNAベクターをplasmid DNAやウイルスを使って細胞に遺伝子導入を行うと、まず核内でshRNAが転写され、①センス鎖(サイレンシングされるmRNAと同じ配列)、②一本鎖ループ、③アンチセンス鎖(センス鎖に相補的な配列)で構成されるヘアピン構造が形成されます。転写されたshRNAは核外へ移行し、細胞質でDicerによるプロセッシングをうけてRNA誘導サイレンシング複合体(RISC)内に取り込まれ、標的mRNAを認識し、分解します(図1)。
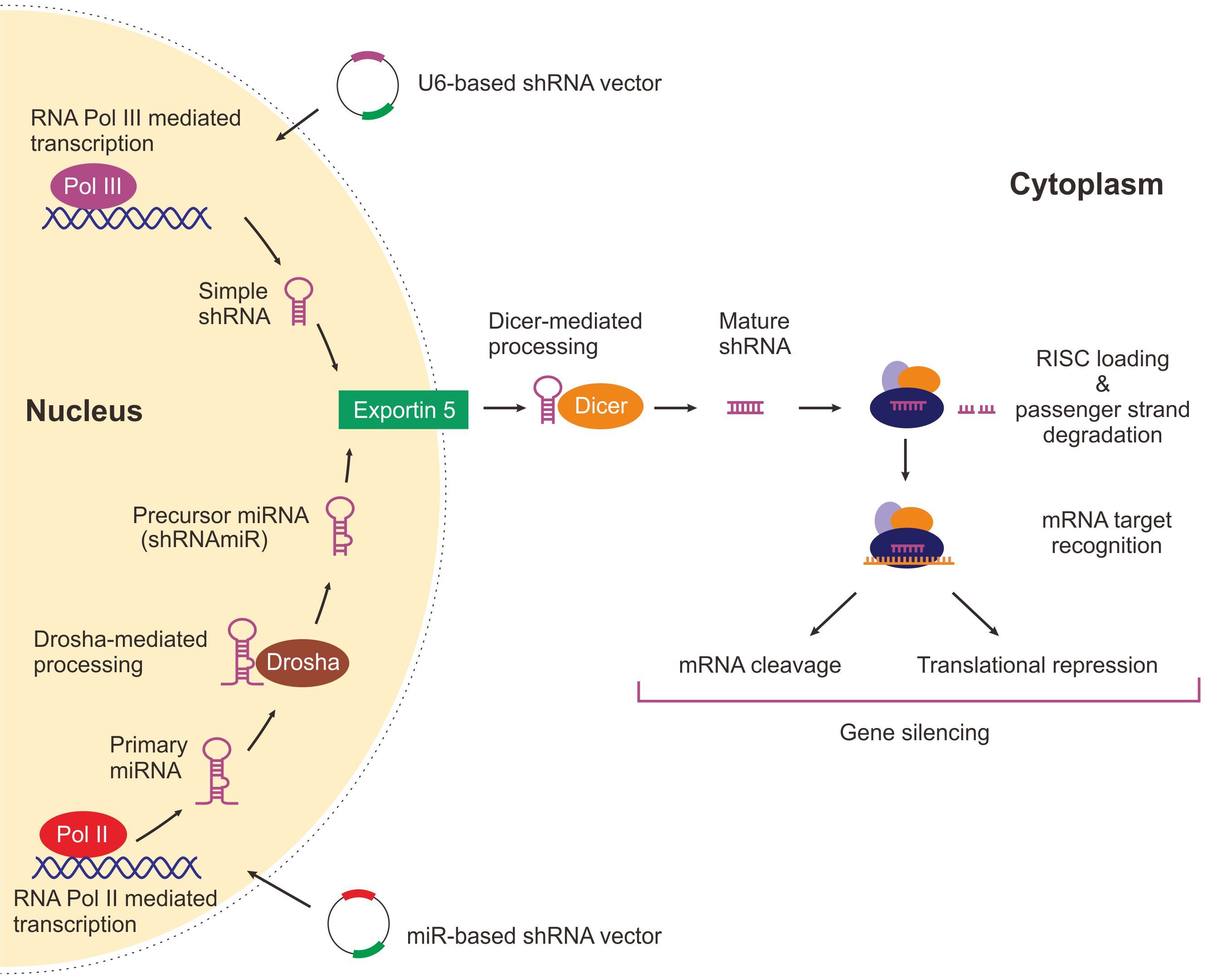
図 1 . U6-ベースと miR-ベースの shRNAを介した遺伝子発現ノックダウン
shRNAを介した遺伝子ノックダウンは、従来のsmall interfering RNA(siRNA)よりも優れた点が多いため、現在では多くの RNAi アプリケーションでshRNAが利用されています。
siRNAを介した遺伝子ノックダウンとshRNAを介した遺伝子ノックダウンの特長を、以下の表にまとめます。
| shRNAを介したノックダウン | siRNAを介したノックダウン | |
|---|---|---|
| デリバリー方法 | 採用するベクターに依存。トランスフェクションまたはトランスダクション | トランスフェクション |
| ノックダウンされる期間 | 長期的 | 一過的 |
|
エピソーマルまたは安定インテグレーション |
デリバリー方法に依存。エピソーマル、または安定インテグレーション | エピソーマル |
| 選択マーカーの追加 | 可能,。蛍光またはドラッグ選択マーカー | 不可 |
| 細胞種 | 幅広い細胞へのアプリケーションに適してる | 高いトランスダクション効率の細胞にのみ適している |
| オフターゲットのリスク | オフターゲットのリスクが低減 | オフターゲットのリスクが高い |
| 分解率 | 低い | 高い |
shRNAの発現を調節するには
shRNA発現ベクターは、U6ベースおよびmiRベースの2タイプがよく使用されています。U6ベースでは、U6プロモーターなどのRNAポリメラーゼIIIプロモーターでステムループ型shRNAを発現させます。一方、miRベースでは、RNAポリメラーゼIIプロモーターで、miR配列を付加したshRNAを発現させます。miR由来の配列はprimary miRNAと同様のメカニズムでプロセッシングされshRNAが生成されます。いずれのベクターから発現させても、細胞内では同様のメカニズムで、最終的にmiRNAにプロセッシングされ、標的遺伝子の発現を抑制します (図 1)。
miRベースのベクターでは、RNAポリメラーゼIIプロモーターを使用するため、任意のプロモーターによって、様々なアプリケーション(組織特異性、発現誘導性、および発現強度)で発現させることが可能です。miRベースのベクターには、この他にも優れた点がいくつかあります。1つ目は、複数のshRNAを1つのポリシストロンで発現できる点です。細胞内プロセッシングによって、単一の転写産物から複数の成熟shRNAを生成させることができるため、同時に複数の遺伝子をノックダウンしたり、同一遺伝子内の複数の領域をターゲットとすることができます。2つ目は、shRNAとタンパク質を1つのポリシストロンで発現できる点です。タンパク質とshRNAの共発現が必要な場合に使用できることに加え、タンパク質にマーカーを選択すれば、shRNAの転写を直接モニターすることも可能です。
miR ベースのベクターではshRNA の発現をフレキシブルに制御することができますが、より強力にノックダウンできるのはU6 ベースのベクターであることが多いようです。したがって、miRベースのベクターを使用する必要がない限り、通常、遺伝子ノックダウン実験には、U6ベースのベクターの使用をお考えください。
下の表にU6ベースのベクターとmiRベースのベクターの特長をまとめています:
| U6ベースshRNAベクター | miRベースshRNAベクター | |
|---|---|---|
| shRNAの構造 | stem-loop shRNA | stem-loop shRNAにmicroRNA スキャフォールドを付加したもの |
| shRNAの長さ | 50-70 nt | >250 nt |
| プロモーター | RNAポリメラーゼ III プロモーター( U6 または H1) | RNAポリメラーゼ II プロモーター(ユビキタス、組織特異的、誘導性プロモーターなど) |
| shRNAのプロセスメカニズム | Dicerによる細胞質でのプロセシング | Droshaによる核内でのプロセシングとDicerによる細胞質でのプロセシング |
| 1ベクターで発現させることのできるshRNAsの数 | 単一のshRNA (一般的な場合) | 単一から複数のshRNA |
| shRNA転写産物からORFタンパク質を共発現させること | 不可 | 可能 |
| 遺伝子ノックダウン効率 | 大変強力な場合が多い | U6ベースより低い場合が多い |
| 細胞毒性 | 高い | U6ベースより低い |
実験による検証
当社のレンチウイルスU6型shRNAノックダウンベクターは図2にあるように、高効率な遺伝子ノックダウンが可能です。ここではU6型とmiR30型shRNAシステムの比較もしています。
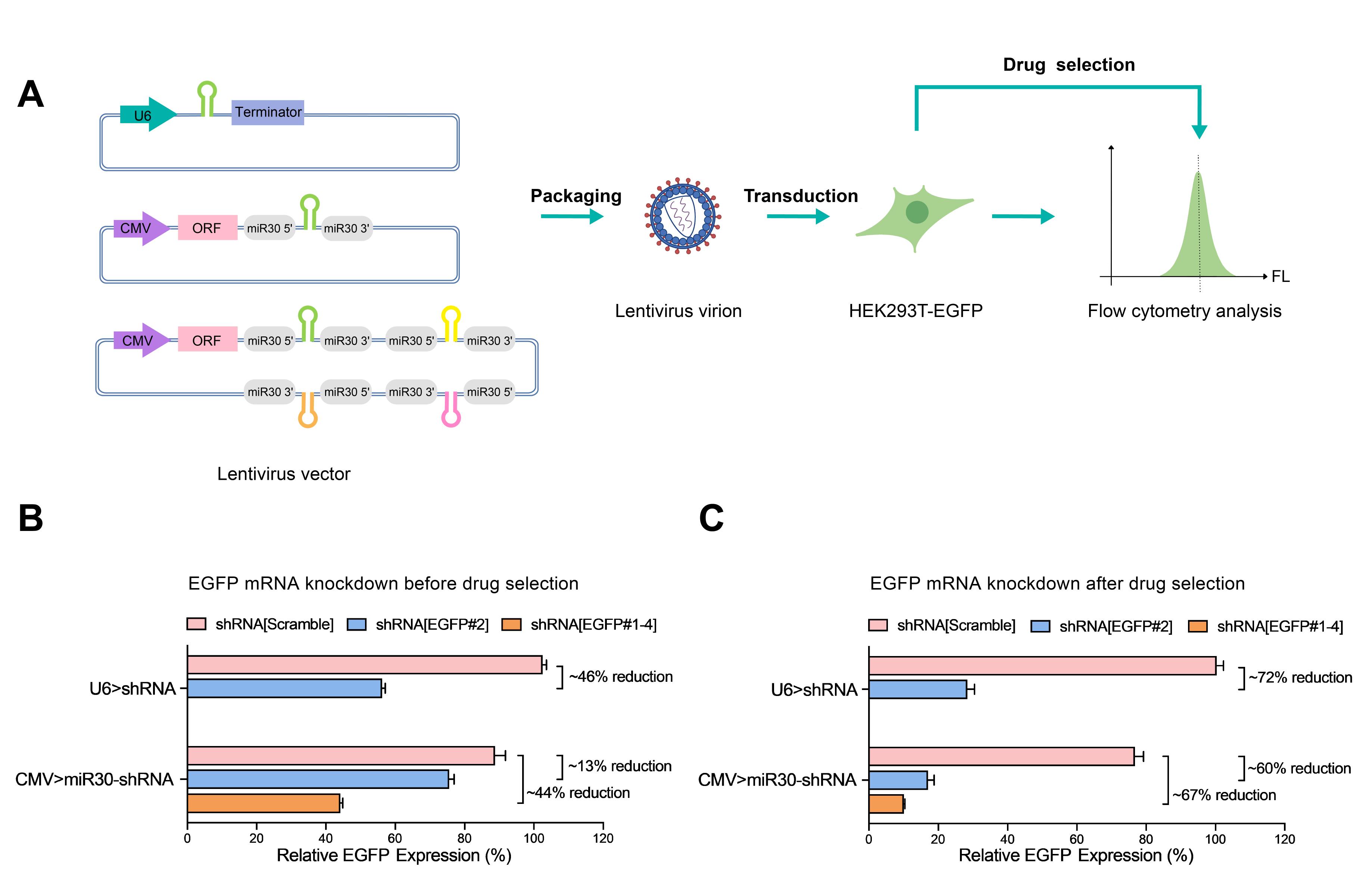
図2 U6型とmiR30型shRNAレンチウイルスシステムを使用した、EGFPノックダウン効率の比較。
(A) U6プロモーターから発現するshRNA、CMVプロモーターから発現するmiR30 shRNA、もしくはCMVプロモーターから発現するmiR30 クアッドshRNAが組み込まれたレンチウイルスベクターをウイルスにパッケージングした。EGFPを発現するHEK293T細胞にレンチウイルスに感染させたのち、薬剤選択の前と後におけるEGFP発現量をフローサイトメトリーによって測定した。(B) 薬剤選択の前のEGFP発現量は、U6型shRNAでは~46%の減少(P<0.001)、CMVプロモーターから発現するmiR30型shRNAで13%の減少(P<0.001)、CMVプロモーターから発現するmiR30型クアッドshRNAで44%の減少(P<0.001)がみられた。(C) 薬剤選択後のEGFP発現量は、U6型shRNAでは~72%の減少(P<0.001)、CMVプロモーターから発現するmiR30型shRNAで60%の減少(P<0.001)、CMVプロモーターから発現するmiR30型クアッドshRNAで67%の減少(P<0.001)がみられた。相対EGFP発現量は感染細胞の蛍光強度の中央値(MFI:median fluorescence intensities)を非感染細胞の中央値で割ることによって計算された。実験は3回繰り返され、標準偏差は図内で記載している。p値はTukey検定から算出した。
shRNAデータベース
VectorBuilderのshRNAベクターデザインツールは、主要な生物種に対して最適化されているshRNAデータベースに直結しています。そのため、目的遺伝子に対して高いノックダウン効率をもつshRNAを簡単にストレスフリーにベクターにデザインすることができます。shRNAsの設計には RNAi コンソーシウムAllと同等のルールを採用しています。スコアーは全て0点以上(≥0)、平均値は~5点、標準偏差は~5、および95%のスコアーは15点以下(≤15)となります。ノックダウンスコアが最高の15点の時、ノックダウン効率は最高で、最低の0点時、ノックダウン効率は最低になります。ベクター構築には、ノックダウンスコアがなるべく15点に近いshRNAを使用します。
VectorBuilderのオンラインベクターデザインプラットフォームでは、遺伝子名を入力するだけで、データベース上にある、すべてのshRNA情報が検索でき、簡単にベクターをデザインすることができます。各shRNAはUCSCゲノムブラウザにリンクされているるため、ゲノム上、およびすべてのトランスクリプトのアイソフォーム上におけるシークエンス情報が確認できます。当社データベースでは、ノックダウンスコアの高い順にshRNAをランク付けしています。初めてノックダウン実験を行うときは、上位3位のshRNAでテストしてみることを推奨しています。しかしノックダウンスコアは、あくまでも目安で、低スコアのshRNAでも高いノックダウン効率が得られる場合もあり、実際のノックダウン効率は、スコアが予測したものとは大きく異なる可能性もあるので注意が必要です。すでに論文で報告され、効果の認められているターゲットシークエンスがある場合は、そちらを使う方が結果への近道です。また、3'UTRをターゲットにしても、コーディング領域をターゲットにした場合と同じ効果が得られる場合もあります。
VectorBuilderのオンライン “リソース” には、shRNAを介したRNAi 実験の計画・実施、さらにトラブルシューティングに関する豊富な情報をご提供しています。
shRNAベクターに関してのガイドを読むshRNAベクターのカスタマイズに参考になるベクターコンポーネントガイドを読む
shRNA関連参考文献
Mol Cell. 9:1327 (2002); miR30ベースの遺伝子ノックダウン
Nucleic Acids Res. 34:e53 (2006); miR155ベースshRNAベクター開発
J Gene Med. 9:620 (2007); IPTG誘導性遺伝子ノックダウンシステム
Proc Natl Acad Sci USA. 101:10380 (2004); Cre-lox-による遺伝子ノックダウンシステムの開発
リソース
shRNAツールに関するQ&A
機能喪失を指標とした遺伝子研究において、shRNAによるノックダウン およびnucleaseによるノックアウト (例 CRISPR やTALEN) は、いずれも優れた方法ですが、実験目的に最適な方法を決定するには、考慮すべき点がいくつかあります。
メカニズム
- ノックダウンベクター:ノックダウンベクターでは、標的mRNAの機能を抑制する短いヘアピンRNA(shRNA)を発現させます。shRNAは、mRNAの切断とその翻訳を抑制することにより、目的遺伝子の機能を抑制します。shRNAノックダウンベクターは、細胞ゲノムをDNAレベルで改変することはありません
- ノックアウトベクター:CRISPRとTALENは、ゲノム中の特定の標的部位を切断するようにヌクレアーゼを誘導し、細胞ゲノムをDNAレベルで改変します。ヌクレアーゼによって切断された部位は、細胞修復機能によって修復されますが、その修復は完全ではないため、修復部位に小さな挿入や欠失などの変異が導入されます。これらの変異導入により、フレームシフトや早期に停止する終始コドンが生じ、その結果、目的遺伝子の機能が喪失されます。ゲノム内、近距離(数kb以内)の2か所を同時に標的とした場合には(例 dual-gRNA)、2か所で挟まれた全領域を欠失させることができます。
効果
shRNAによるノックダウンでは、標的遺伝子の発現を完全に抑制することはできず、最も効果的なshRNAでも、発現の一部は残ってしまいます。一方、CRISPRおよびTALENでは、長期安定的な変異を導入できるため、完全に遺伝子機能を喪失させることが可能です。
安定性と均一性
shRNAベクターは、導入細胞間でのばらつきは少なく、実験間で非常に安定した結果が得られます。一方、CRISPRやTALENでは、変異の導入は確率に依存するため、細胞間のばらつきが非常に大きくなります。また、目的遺伝子を完全にノックアウトするには、細胞内すべての遺伝子コピーをノックアウトする必要があります。正常な細胞では、1遺伝子に対し2つのコピーが存在しますが(XまたはY関連遺伝子を除く)癌細胞では2つ以上のコピーが存在する場合があります。複数の遺伝子コピーが存在する場合、すべての遺伝子コピーをノックアウトすることは難しく、すべての遺伝子コピーがノックアウトされていることを確認するには、シークエンスによるクローンのスクリーニングが必要になります。
オフターゲット効果
オフターゲット効果は、shRNAによるノックダウン、およびヌクレアーゼによるノックアウトの双方で報告されています。shRNAにおけるオフターゲット効果は、1つの遺伝子に対して複数の異なるshRNAを使用することで検証することができます。複数の異なるshRNAで、一貫して同じ表現型が得られれば、オフターゲット効果による可能性を排除することができます。CRISPRまたはTALENにおけるオフターゲット効果は、複数のクローンで表現型を分析し、検証する必要があります。オフターゲット部位はバイオインフォマティクス的手法で同定することも可能です。
相同組み換え用のドナーベクターをデザインする全てのshRNAが効果的にワークするわけではありません
VectorBuilderの経験と、ユーザーからのフィードバックを総合すると、一般的にある遺伝子に対して 3 ー4 種類のshRNAを試した場合、 2-3種類のshRNAで満足いくノックダウン効果が得られていることがわかっています。しかし選択した全てのshRNAがワークするわけではないことは必ず念頭に置いてください。一般的に、~50-70% のshRNAがノックダウンの表現型を表し、そのうちの ~20-30% は強いノックダウン効果を表します。仮に1-2種類のターゲットshRNAシークエンスを特定の1遺伝子に試した場合、チャンスとしては満足いくノックダウン効果が観察されないこともあり得ます。その場合、検証された別のshRNAを試すことが勧められます。複数の異なるshRNAをミックスした“カクテル” shRNAを試す場合もあり、ノックダウン効率の改善に役立つこともあります。
目的遺伝子のノックダウン効果を検証するアッセイ系が正しく実施できていない
最も一般的で感度の高いshRNAノックダウンの評価アッセイ系はRT-PCRです。複数のプライマーペアーを試し、最も特異性の高いプライマーペアーを使うことをお勧めします。一般的に、RT-qPCRのプライマーは、exon-exonのジャンクションシークエンスを挟むことで、サンプルにコンタミしたゲノム由来のDNAからの増幅を避けるようにデザインします。新しいプライマーペアーを試す際は、アガロースゲル電気泳動でバンドサイズを確認したり、サンガーシークエンスで増幅したPCR産物の確認を行うこともあります。RT-PCRには、必ずminus-RTコントロールを取って、ゲノムDNAのコンタミ率を概算できるようにしてください NCBI primer designing tool はデザインしたプライマーをコンピューター上(in silico)で検証できる良いツールです。
ウエスタンブロッティングもノックダウン効率検証のよいアッセイ系です。弱点は使用する抗体によっては非特異的な偽陽性バンドが出てしまい、結果を見誤るリスクがあることです。ウエスタンブロッティングに使う抗体は、目的遺伝子産物を特異的に認識することを予め確認してから使用してください。
shRNAが目的遺伝子の転写アイソフォームの一部にしかターゲッティングできていない
shRNAをデザインする際、目的遺伝子にisoformが存在する場合は、できる限り多くのisoformをターゲットできるようにデザインすることをお勧めします。 反対に、特定のisoformのノックダウンでは、可能な限り特異性の高いターゲットを選択してください。VectorBuilderは、特定の動物種に対して、最適化したshRNAデータベースを確立しています。shRNAベクターをご自分でデザインし、VectorBuilderに受託構築をご依頼いただく際は、VectorBuilderのデータベースをお試しになられることをお勧めします。このデータベースには、全てのshRNAのリスト、シークエンス、スコアーが一覧表示され、かつUCSC Genome Browserへのリンクからゲノムシークエンスのどの位置にターゲットシークエンスが位置するか確認することができます。
VectorBuilderのshRNAは the RNAi consortium (TRC) のアルゴリズムをベースとして改良したshRNAデザインとスコアーを採用しています。各RefSeq に登録されている転写産物に対して、ターゲットサイトの候補となる21merシークエンスが全て同定されています。候補ターゲットサイトは、ノックダウン効率や特異性を低下させるような特長が検出されたり、クローニング不可性 (同一塩基≥4 連続、G または C塩基の≥7連続、GC 含量<25% またはGC含量>60%、そして 5’末端にAA )が検出された場合、候補から除外されます。ノックダウンスコアーは、内部にステムループ構造を持つ、3’末端に向てGC含量が高くなる、既知の miRNA シードシークエンス、他の遺伝子に対するオフターゲットマッチなどが検出されるとペナルティーをつけ、スコア化しています。また、1遺伝子に対して複数の転写産物が同定されている場合(alternative transcripts)、全ての転写産物に対して、最も高いスコアーを出すターゲットサイトを選び出しています。
全てのスコアーは ≥0以上の数字で、中間値(mean)が ~5、そして標準偏差が ~5となっています。95% のshRNAでスコアーは≤15となっています。例えば、あるshRNAのノックダウンスコアーが15であるとすると、最も効果の高いノックダウンパフォーマンスが予想され、クローニングも可能と判断されます。一方、ノックダウンスコアーが0のshRNAでは、最悪のノックダウンパフォーマンス、またはクローニング不可能なターゲットシークエンスと判断されます。
ただ、ノックダウンスコアーはあくまでも予測シークエンスです。実際のノックダウン効率はスコアーからかけ離れている場合もあるかもしれません。低いノックダウンスコアーが与えられたターゲットシークエンスでも、shRNAとして研究論文につかわれていて、比較的よくワークしている場合もあります。また、3'UTR領域のターゲットでも、コーディング領域のターゲットの場合と同様に効果があることも念頭に置いてください。